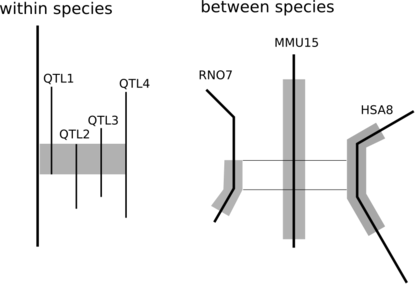
Genetics researchers have long utilized QTL and association studies to try and pinpoint those parts of the genome that underlie disease or production phenotypes. A wealth of papers exists describing these results, resulting in the opportunity to fine-map QTL regions. If several papers describe a QTL for the same trait and those QTLs overlap with each other, it is the overlap between these QTLs that is likely to contain the gene of interest.
Unfortunately, even though much data is ‘out there’, it cannot be fully used. The current practice for doing QTL and association studies is presented in the figure below. Basically, wet-lab research (genotyping and phenotyping) is followed by a statistical analysis to identify QTLs or associated markers and results are published in a paper. Even though the paper in itself can be valuable, the data and results embedded in it can be hard to extract. In addition, QTL and association database curators have to mine literature and identify those papers that hold relevant information.
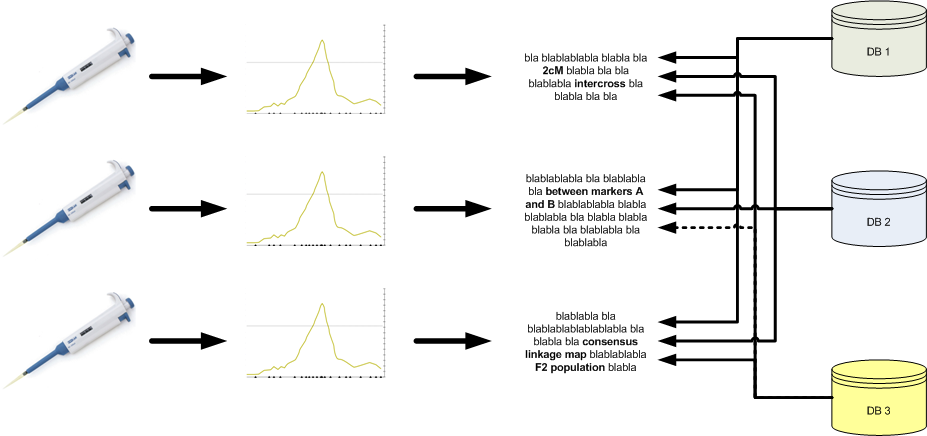
In addition to the issues involved with this practice, the fact that no minimum standards are set results in highly inconsistent reporting. Some of the issues include:
The MIQAS set of rules accompanied with the standardized XML and tab-delimited file formats will serve two goals:
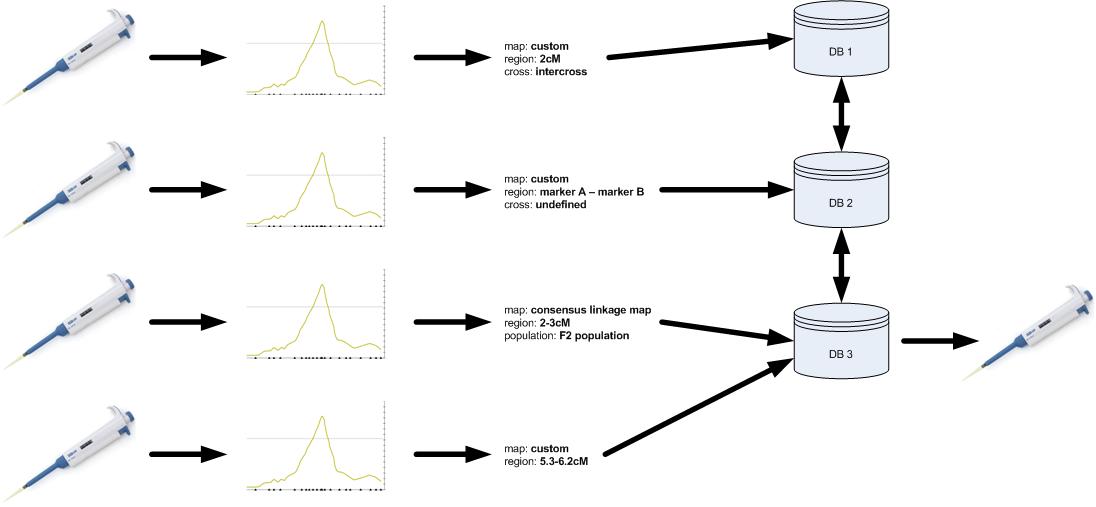
Several research communities have already made a similar step to make the above happen. Good examples are the publication of results for microarray and SNP discovery efforts. If a researcher wants to get a paper published that describes the discovery of new polymorphic markers (e.g. SNPs), it is often no longer possible to get away with listing the PCR primers in a long table within the manuscript. More and more, journals request that those markers are first submitted to a central database such as dbSNP and that only the accession numbers of the markers are mentioned in the paper. Even though this involves a little bit of added work for the scientist, the added value of having the markers available through dbSNP outweighs this disadvantage by far. The microarray community uses the same approach with the MIAME standard.
Using MIQAS, the QTL and association research community can start to employ a similar paradigm for reporting their results. Before submitting the manuscript to a journal, the research group first submits the data and results to a MIQAS-compliant database. This database creates an accession number for the study that can be referenced in the manuscript.
To make a workflow as described above (similar to microarry results) work, several parties have to play a role in the process:
The central thing described in a MIQAS_XML file is a QTL or association study. This means that if a study results in more than one QTL, all of these can be described together in one MIQAS file.
Apart from general information about the QTL/association analysis like a name, description and reference, most of the information refers to (1) the trait that was measured, (2) the experiment that was used and (3) the associated markers or positions of any QTLs.
The MIQAS_TAB specification outlines the format for a group of tab-delimited files to support submission and exchange of MIQAS-related data. This is your primary source of information about what aspects of a QTL or association study have to be recorded.
An XML Schema (called MIQAS_XML and available here) is available for data exchange between MIQAS-compliant databases. This file format is also to be used to submit data to a MIQAS-compliant database. For an example of such a file, see here. As there’s a one-to-one mapping with the MIQAS_TAB specification, please see there for more information.
There are two basic ways that researchers can get their data and results in a MIQAS XML file:
You can also create a group of tab-delimited files according to the MIQAS_TAB specification. A website will be created (but not available yet) that can convert these files into a single MIQAS_XML file.
Please send inquiries to jan.aerts@kuleuven.be or jreecy@iastate.edu